
Systemic amyloidosis — extra information
Introduction
Demographics
Causes
Clinical features
Complications
Diagnosis
Differential diagnosis
Treatment
Prognosis
Systemic AA amyloidosis
What is systemic amyloidosis?
Systemic amyloidosis is an uncommon disorder in which misfolded proteins become resistant to normal processes of removal by the body. Their accumulation in bodily organs leads to abnormal organ function.
The heart, liver, kidneys, nerves, lungs, and bowel may be affected, and in only one type of systemic amyloidosis (AL type) is the skin significantly affected and indeed may be the clue to discovering underlying internal organ disease.
Systemic amyloidosis is subclassified according to the chemical composition of the misfolded protein. There are two components to the amyloid:
- A constant unit, most commonly amyloid P component
- A variable unit such as:
- Immunoglobulin light chain (producing 'AL' amyloid)
- Transthyretin (producing ATTR amyloid)
- Beta 2 microglobulin (Aβ2M) which produces a systemic amyloidosis in chronic dialysis.
Chronic inflammation due to autoinflammatory conditions, autoimmune disease, and infections may induce AA systemic amyloid (see section below). This does not produce skin signs or symptoms but may be identified histologically in subcutaneous fat.
For information on localised cutaneous amyloidosis, click here.
Who gets cutaneous features of systemic amyloidosis?
Cutaneous manifestations only arise from the AL type (amyloid light chain), which is associated with plasma cell dyscrasias, such as myeloma. The incidence is 12 cases per million patients a year, and affects older age groups without racial or gender bias.
Overall worldwide, AA amyloidosis outnumbers all other forms due to high prevalence of infectious diseases in the developing countries.
What causes the general features of systemic amyloidosis?
Extracellular deposition of insoluble amyloid protein in affected organs as well as the skin and its adnexae causes tissue destruction leading to dysfunction. However, the precise mechanism of how amyloid fibril is formed and sequestrated into tissues has not been well understood yet.
What are the clinical cutaneous features of systemic amyloidosis?
Cutaneous manifestations will vary depending on the site of amyloid deposition and the degree of local tissue destruction; it occurs in 30–40% of patients with primary systemic AL amyloidosis.
Usual features include:
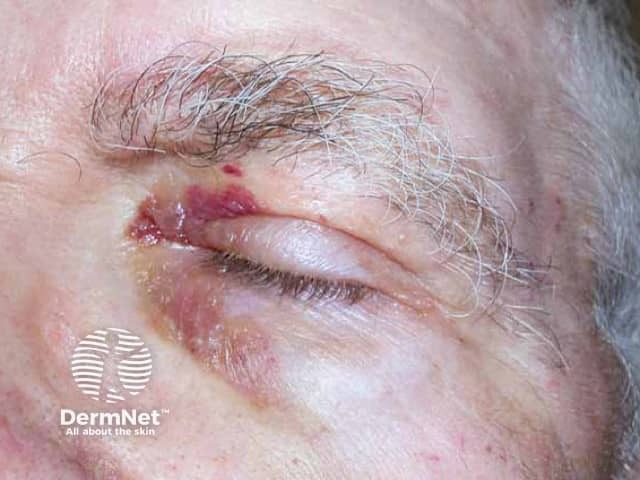
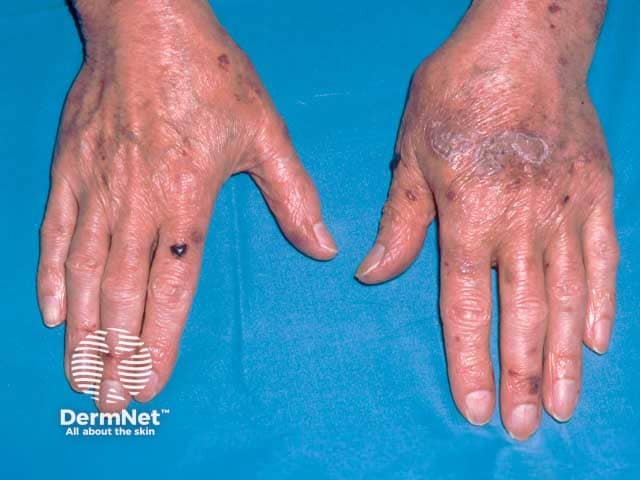
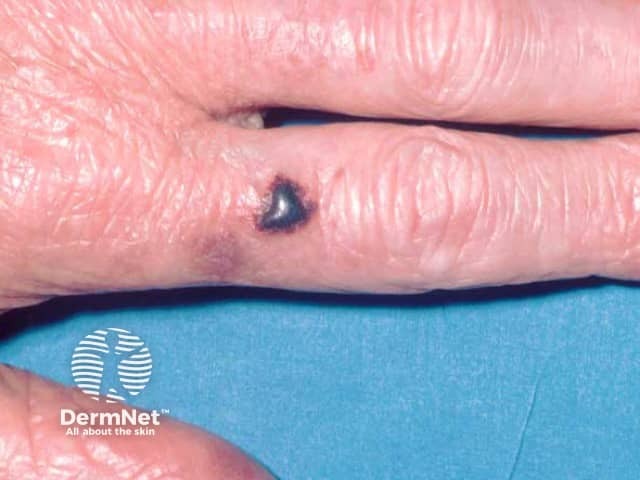
- Purpura, ecchymoses, petechiae, and haemorrhagic blisters especially over eyelids and the periorbital areas, due to capillary wall fragility from amyloid infiltration of vessel wall
- Can be triggered by minor trauma and even coughing
- Papules, plaques, or nodules with a shiny-waxy or haemorrhagic appearance in intertriginous areas such as eyelids, retro-auricular, neck, axillae, and the anogenital region
- Hair loss (alopecia) from destruction of hair caused by amyloid deposit around the pilosebaceous unit
- Nail dystrophy (ridging, splitting and brittleness of nail plate) from amyloid deposition in the nail matrix
- Scleroderma-like skin including a thickened folded scalp
- Coarse facies with loss of wrinkles
- Macroglossia (diffusely enlarged and firm tongue) from amyloid infiltration
- Xerostomia (dry mouth) from destruction of salivary glands
- Peripheral nerve thickening, producing neuropathy and carpal tunnel syndrome
- Organomegaly such as enlargement of liver, heart, and kidneys, leading to eventual organ failure
- There can also be general symptoms of weight loss, weakness or fatigue, dyspnoea, oedema, paraesthesia, lightheadedness, syncope, constipation or diarrhoea, and hoarse voice.
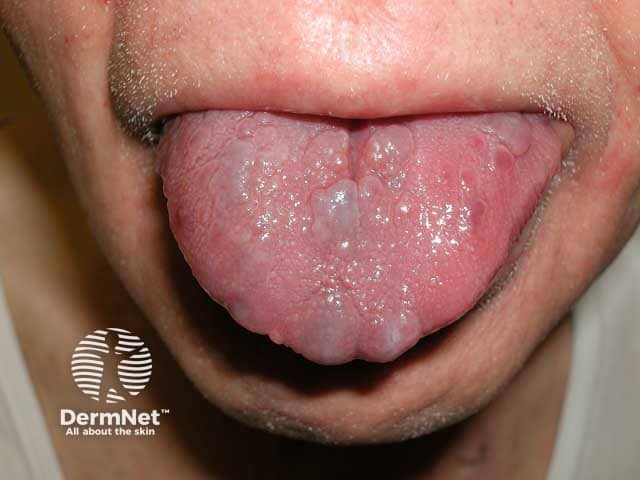
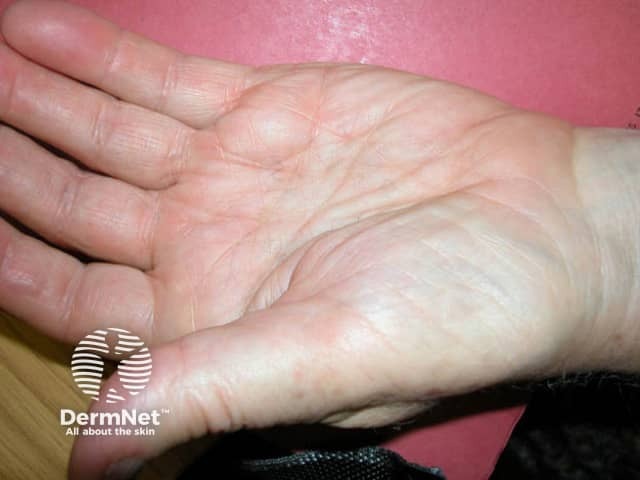
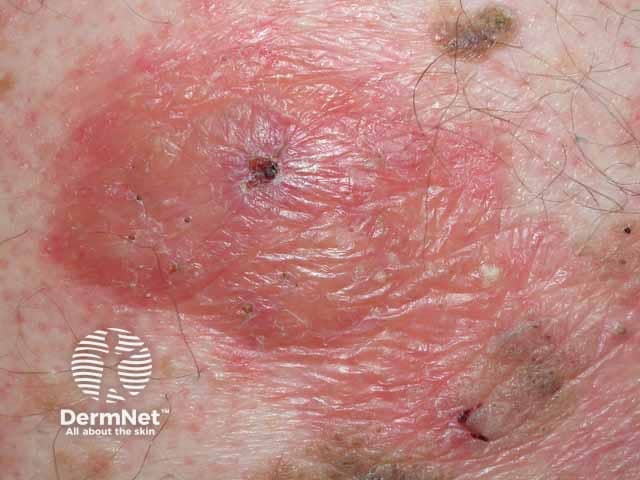
What are the complications of AL systemic amyloidosis?
The complications are variable depending on the site and extent of involvement.
- Skin, hair, and nail involvement can lead to disfigurement and psychological distress.
- Macroglossia can cause chewing and swallowing problems.
- Organ failure and death (in particular renal failure, nephrotic syndrome, and cardiomyopathy).
How is systemic AL amyloidosis diagnosed?
- Blood tests including full blood examination, liver and renal function, and urinalysis.
- Serum (immuno) electrophoresis, serum free light chain assay, urinary Bence Jones protein to identify associated paraprotein.
- Skin biopsy is generally required to identify and classify the amyloid.
- H & E staining will show amorphous eosinophilic deposits from the upper dermis to subcutaneous structures.
- Congo-red stain with +/- immunofluorescence (apple-green birefringence with polarized light) will confirm amyloid deposit.
- Immunohistochemical analysis will identify specific types of amyloid protein (eg, AL, AA, or others).
- The extent of internal organ involvement in AL amyloid can be visualised by radioisotope serum amyloid P component scanning available in specialised centres.
What is the differential diagnosis for systemic AL amyloidosis?
The differential diagnosis will depend on presenting features, and include:
- Several chronic dermatoses including scleroderma, cutis verticis gyrata, and pseudoxanthoma elasticum from the appearance of red-brown discoloured or thickened skin.
- Bleeding disorders and scurvy for purpura, petechiae, and ecchymosis of skin.
- Various cutaneous neoplasms for the nodular subtype.
- Various hair and nail disorders for hair loss and nail dystrophy.
What is the treatment of systemic AL amyloidosis?
Frequently used
- Conventional high dose plasma cell directed chemotherapy with melphalan and dexamethasone.
- Sometimes followed by (in those well enough to tolerate it) autologous stem cell transplant.
Alternatives
- Daratumumab, a monoclonal anti-CD 38 antibody, targets this antigen present on plasma cells and is a newer alternative.
- Bortezomib, an inhibitor of the 26S proteasome, induces apoptosis of actively proliferating cells and is also showing some promise.
What is the prognosis for AL systemic amyloidosis?
The average time from identification of systemic AL amyloidosis to death is several months if left untreated. Some treatments, including stem cell transplantation and chemotherapy, have achieved 10-year survival periods.
Cardiac and renal amyloidosis are poor prognostic features.
Which skin conditions produce systemic AA amyloidosis?
Chronic inflammation of the skin can rarely induce systemic AA amyloidosis, and chronic pustular psoriasis and psoriatic arthritis are perhaps the best established causes.
The chronic autoinflammatory disorders TNF receptor periodic syndrome (TRAPS) and cryopyrin-associated periodic syndrome such as Muckle-Wells syndrome, familial cold autoinflammatory syndrome (familial cold urticaria), chronic infantile neurological cutaneous and articular syndrome, and hyperimmunoglobulin IgD syndrome all have their own cutaneous features, and can induce systemic AA amyloidosis (which does not show any skin features per se).
Bibliography
- Flores-Bozo LR, Echevarría-Keel J, et al. Mucocutaneous manifestations in systemic amyloidosis A retrospective analytical study in a tertiary care center. Int J Dermatol. 2019 Sep;58(9):1062–8. Journal
- Gertz MA, Dispenzieri A. Systemic Amyloidosis Recognition, Prognosis, and Therapy: A Systematic Review. JAMA. 2020;324(1):79–89. doi:10.1001/jama.2020.5493. Review
- Kumar S, Sengupta RS, Kakkar N, Sharma A, Singh S, Varma S. Skin involvement in primary systemic amyloidosis. Mediterr J Hematol Infect Dis. 2013;5(1):e2013005. Journal
- Lachmann HJ. Periodic fever syndromes. Best Pract Res Clin Rheumatol. 2017;31(4):596–609. doi:10.1016/j.berh.2017.12.001. Journal
- Marzano AV, Damiani G, Genovese G, Gattorno M. A dermatologic perspective on autoinflammatory diseases. Clin Exp Rheumatol. 2018;36 Suppl 110(1):32–8. PubMed
- Picken MM. The Pathology of Amyloidosis in Classification: A Review. Acta Haematol. 2020;143(4):322–34. Journal
- Real de Asúa D, Costa R, et al. Systemic AA amyloidosis: epidemiology, diagnosis, and management. Clin Epidemiol. 2014 Oct 29;6:369–77. Journal
On DermNet
- Systemic amyloidosis case 1
- Cutaneous amyloidosis
- Macular amyloidosis
- Amyloidosis images
- Systemic amyloidosis pathology
- Skin signs and systemic disease
- Scleroderma
- Hair loss
- Nail dystrophy
- Intralesional steroid injections