
Familial cold autoinflammatory syndrome — extra information
Introduction Demographics Causes Clinical features Complications Diagnosis Differential diagnoses Treatment Outcome
What is familial cold autoinflammatory syndrome?
Familial cold autoinflammatory syndrome (FCAS) is a rare autosomal dominant autoinflammatory disease that presents with episodic rash, fever, arthralgias, and fatigue upon generalised exposure to the cold.
FCAS is also known as familial cold urticaria or familial polymorphous cold eruption. It belongs to the spectrum of cryopyrin-associated periodic syndromes (CAPS) or NLRP3-associated autoinflammatory diseases (NLRP3-AIDS), along with Muckle-Wells syndrome (MWS) and neonatal onset multisystem inflammatory disease (NOMID) / chronic infantile neurological cutaneous and articular syndrome (CINCA).
FCAS is the mild phenotype compared to MWS (moderate phenotype) and NOMID/CINCA (severe phenotype).
Who gets familial cold autoinflammatory syndrome?
FCAS is inherited in an autosomal dominant pattern. Children of an affected parent have a 50% chance of inheriting the disease.
Onset of symptoms has been reported from birth. The mean age of presentation is 47 days old, and 95% of affected individuals present by 6 months of age. In rare cases, symptom onset may occur during young adulthood. FCAS is a lifelong condition.
FCAS is extremely rare with an estimated prevalence of 1:1,000,000. It is primarily reported in families of North American and European descent and is seen equally in females and males.
What causes familial cold autoinflammatory syndrome?
FCAS results from a gain of function mutation in the NLRP3 gene which encodes cryopyrin on chromosome 1q44. This leads to the activation of the cryopyrin inflammasome, causing uncontrolled inflammation through the inappropriate release of cytokines including interleukin 1-beta (IL-1β). This results in the inflammatory symptoms of FCAS.
Generalised cold exposure is a trigger for the symptoms of FCAS. The mechanism of temperature dependence is still being studied, but monocytes in patients with FCAS showed enhanced release of cytokine IL-1 on exposure to subnormal temperatures.
What are the clinical features of familial cold autoinflammatory syndrome?
FCAS is characterised by acute, episodic attacks of fever with rash following generalised exposure to cold. The average delay from cold exposure to onset of symptoms is 2.5 hours. Episodes usually last less than 24 hours (on average ~12 hours) and settle spontaneously.
Clinical features of an acute episode may include:
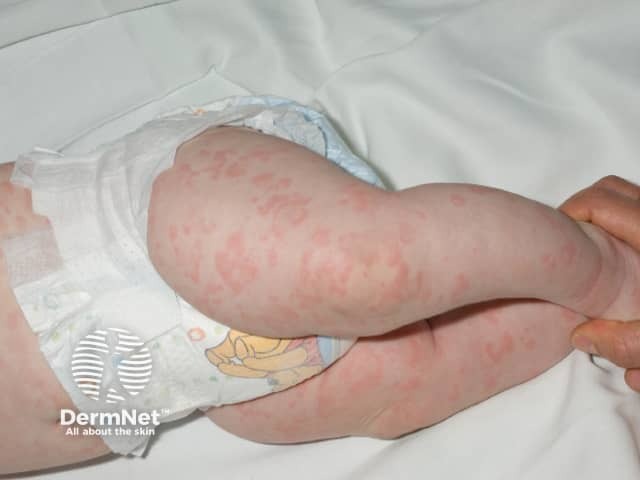
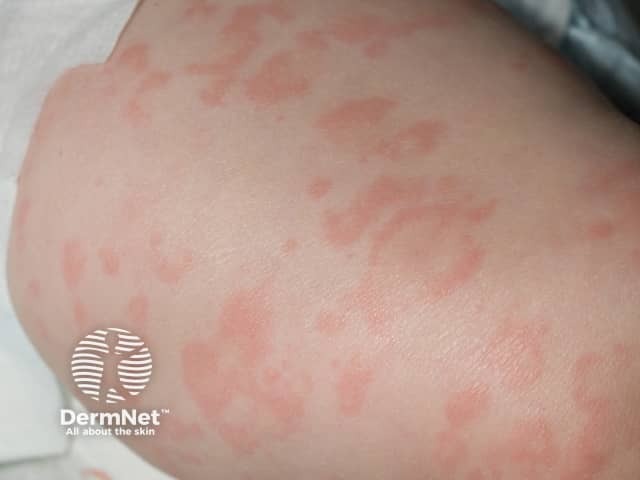
- Rash (100%):
- Often the first sign of FCAS, appearing soon after birth or in early infancy
- While traditionally described as urticaria-like, it is often a non-urticarial, non-pruritic, red maculopapular rash with faint pink figurate patches
- Petechiae have also been described
- The rash often presents first on the extremities alone (45% of cases), the extremities and face (30%), and the extremities and buttocks (11%), followed by generalisation to the rest of the body in most patients.
- Fever and chills (93%)
- Arthralgia and myalgias (96%)
- Conjunctivitis (84%)
- Other reported symptoms:
- Sweating
- Drowsiness
- Headache
- Thirst
- Nausea.
What are the complications of familial cold autoinflammatory syndrome?
- Amyloidosis is a rare complication that occurs in less than 2% of patients with FCAS and can lead to end-stage renal failure.
- The lifelong occurrence of episodic attacks can affect daily activities, mental health, and quality of life.
How is familial cold autoinflammatory syndrome diagnosed?
Diagnosis of FCAS is primarily clinical, based on a history of episodic febrile attacks and rash following generalised cold exposure.
Due to the autosomal dominant inheritance pattern, diagnosis can be accelerated by monitoring children with a known family history of FCAS.
Genetic testing can be performed by NGS (next-generation sequencing) or Sanger sequencing to detect mutations in the NLRP3 gene.
Laboratory findings may include:
- Chronic leukocytosis
- Acute increase in serum interleukin 6 (IL-6) levels and neutrophils (neutrophilia) during a flare
- Elevated C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR).
Skin biopsy shows dermal oedema with neutrophil infiltration in the dermis, particularly perivascular and peri-eccrine.
What is the differential diagnosis for familial cold autoinflammatory syndrome?
- Acquired cold urticaria — often confused with FCAS, although they are distinct syndromes; in cold urticaria, the ice cube test is positive (this is negative in FCAS)
- Familial Mediterranean fever
- Hyperimmunoglobulinaemia D with periodic fever syndrome
- Tumour necrosis factor receptor-associated periodic syndrome (TRAPS)
- Other more severe phenotypes of cryopyrin-associated periodic syndromes (CAPS):
- Muckle-Wells syndrome (MWS) — flares last longer (1–3 days) than FCAS and amyloidosis is more common (up to 30%)
- Neonatal onset multisystem inflammatory disease / chronic infantile neurological cutaneous and articular syndrome (NOMID/CINCA) — more systemic and severe symptoms than FCAS with central nervous system involvement (eg, development delay, seizures, and sensorineural hearing loss).
What is the treatment for familial cold autoinflammatory syndrome?
General measures
- Adequate rest and keeping warm.
- Moving to more temperate climates to reduce the frequency of attacks.
- Non-steroidal anti-inflammatory drugs and glucocorticosteroids during flares.
Specific measures
Interleukin 1 (IL-1) blocking therapy is now the recommended standard of care for FCAS. These biological treatments block the effect of IL-1β on the IL-1 receptor, inhibiting downstream signalling which reduces systemic inflammation and improves symptom control. Dosing requirements may vary depending on severity of disease.
Currently available IL-1 blockers (given by subcutaneous injection) include:
- Canakinumab (human monoclonal antibody binding to IL-1β)
- FDA-approved for adult and paediatric patients over 4 years of age (2009)
- Paediatric dosing: 2–8 mg/kg 8 weekly
- Adult dosing: 150–600 mg 8 weekly.
- Rilonacept (recombinant fusion protein binding to IL-1a and IL-1 )
- FDA-approved for adults and children 12 years and older (2008)
- Paediatric dosing: 4.4 mg/kg loading dose and 2.2 mg/kg weekly maintenance dose
- Adult dosing: 320 mg loading dose and 160 mg weekly maintenance dose.
-
Anakinra (recombinant IL-1 receptor antagonist)
- Approved in the EU for all forms of CAPS (2013), and in Australia for patients with CAPS aged ≥8 months with a body weight of ≥10 kg (2014)
- Dosing: 1–2 mg/kg daily (up to 8 mg/kg daily for more severe forms of CAPS).
What is the outcome for familial cold autoinflammatory syndrome?
FCAS is a lifelong condition with good long-term prognosis. Patients can be monitored for the rare development of amyloidosis and renal failure by monitoring for proteinuria and microalbuminuria. While the episodic attacks of FCAS can affect quality of life, treatment with IL-1 blockers has greatly improved outcomes for patients with cryopyrin-associated periodic syndromes (CAPS).
Bibliography
- Booshehri LM, Hoffman HM. CAPS and NLRP3. J Clin Immunol. 2019;39(3):277–286. doi: 10.1007/s10875-019-00638-z. Journal
- Hoffman HM, Wanderer AA, Broide DH. Familial cold autoinflammatory syndrome: phenotype and genotype of an autosomal dominant periodic fever. J Allergy Clin Immunol. 2001;108(4):615–620. doi: 10.1067/mai.2001.118790. Journal
- Raghawan AK, Ramaswamy R, Swarup G. Cold-induced loss of interaction with HSC70 triggers inflammasome activity of familial cold autoinflammatory syndrome-causing mutants of NLRP3. Biochem Biophys Res Commun. 2023;641:42–49. doi: 10.1016/j.bbrc.2022.12.018. Journal
- Romano M, Arici ZS, Piskin D, et al. The 2021 EULAR/American College of Rheumatology points to consider for diagnosis, management and monitoring of the interleukin-1 mediated autoinflammatory diseases: cryopyrin-associated periodic syndromes, tumour necrosis factor receptor-associated periodic syndrome, mevalonate kinase deficiency, and deficiency of the interleukin-1 receptor antagonist. Ann Rheum Dis. 2022;81(7):907–921. doi: 10.1136/annrheumdis-2021-221801. Journal
- Rosengren S, Mueller JL, Anderson JP, et al. Monocytes from familial cold autoinflammatory syndrome patients are activated by mild hypothermia. J Allergy Clin Immunol. 2007;119(4): 991–996. doi: 10.1016/j.jaci.2006.12.649. Journal
On DermNet
- Autoinflammatory syndromes
- Periodic fever syndromes
- Cryopyrin-associated periodic syndromes
- Muckle-Wells syndrome
- NOMID/CINCA syndrome
- Systemic amyloidosis
- Biological treatments
- Anakinra
- Cold urticaria
Other websites
- Familial cold autoinflammatory syndrome — Genetic and Rare Diseases Information Centre (GARD)
- Familial cold autoinflammatory syndrome — OMIM