
Epidermolytic ichthyosis — extra information
Introduction Causes Demographics Clinical features Complications Diagnosis Differential diagnoses Treatment Outcome
What is epidermolytic ichthyosis?
Epidermolytic ichthyosis is one of the five main types of ichthyosis along with lamellar ichthyosis, ichthyosis vulgaris, congenital ichthyosiform erythroderma, and X-linked ichthyosis.
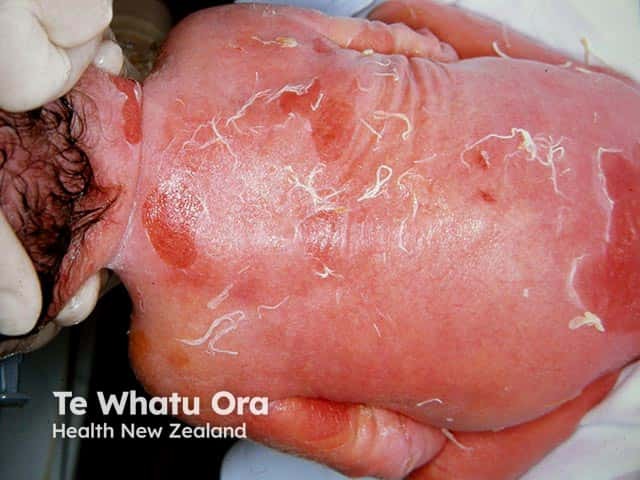
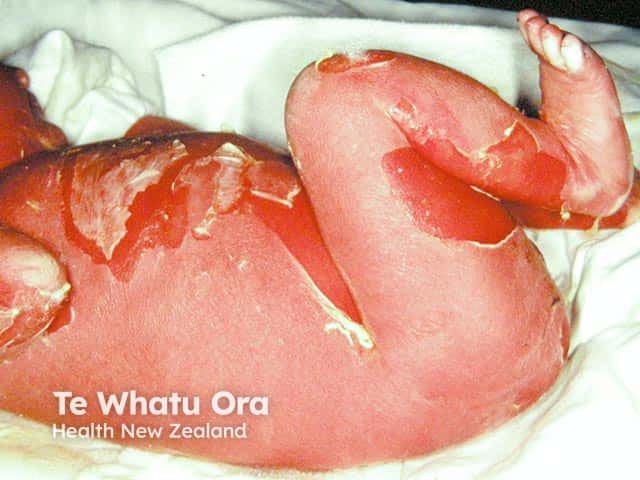
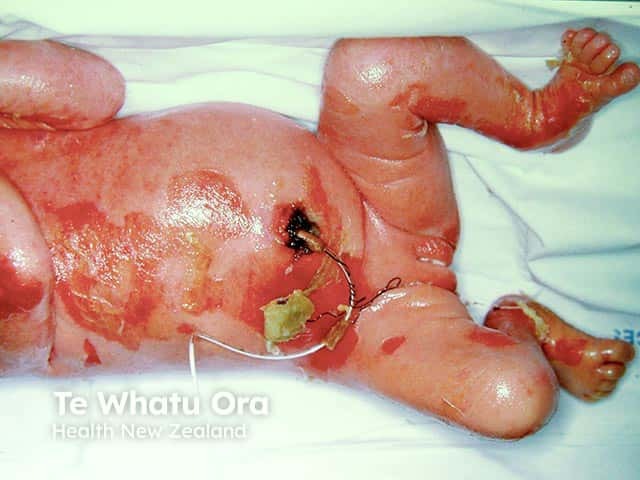
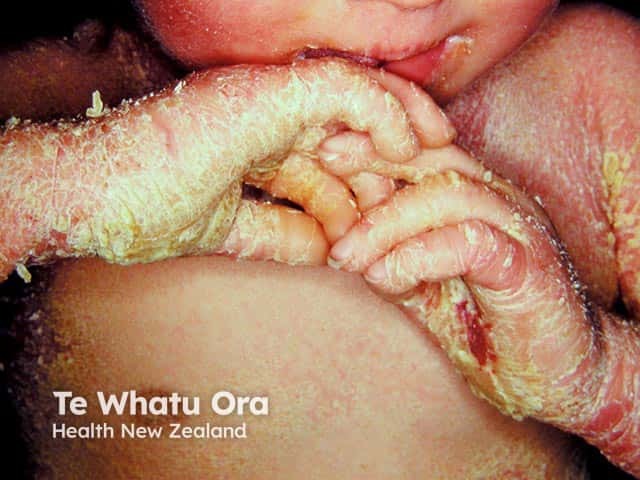
It typically presents at birth with erythroderma, skin fragility, and blistering [1–3].
Epidermolytic ichthyosis was formerly known as epidermal hyperkeratosis and bullous congenital ichthyosiform erythroderma.
Epidermolytic ichthyosis in a neonate
What causes epidermolytic ichthyosis?
Epidermolytic ichthyosis is caused by missense mutations (where a single nucleotide is changed) in the keratin genes keratin 1 (KRT1) [4,5] and keratin 10 (KRT10) [5–7].
- Mutations in KRT1 are associated with severe palmoplantar keratoderma.
- Mutations in KRT10 generally lack palmoplantar symptoms [8,9].
- Epidermolytic palmoplantar keratoderma limited to the palms and soles has mutations in the KRT9 gene [8,9].
Keratin tonofilaments consist of keratin 1 (K1) and 10 (K10) and are a robust structural component of the flattened squamous keratinocytes in the superficial epidermis [10–12].
Mutations in KRT1 and KRT10 lead to variable disruption and decreased stability of the K1/K10 tonofilaments and hyperkeratosis due to lack of desquamation [13,14].
Who gets epidermolytic ichthyosis?
Epidermolytic ichthyosis is a rare disorder seen to affect 1 in 100,000–300,000 infants in males and females equally [1–3].
- Spontaneous mutation occurs in 50% of cases [1,2].
- It can have an autosomal dominant mode of inheritance with complete penetrance.
- Autosomal recessive epidermolytic ichthyosis with loss-of-function KRT10 mutations has been reported in three consanguineous families [15–18].
Epidermolytic ichthyosis is the only keratin disease associated with genetic mosaicism; the offspring of parents with epidermolytic epidermal naevi can develop generalised epidermolytic ichthyosis if the gonads are involved [17,19].
What are the clinical features of epidermolytic ichthyosis?
The clinical features of epidermolytic ichthyosis are [1–3]:
- Widespread erythroderma, blisters, and superficial ulceration at birth
- Peeling, erosions, and denuded skin after minor friction or trauma
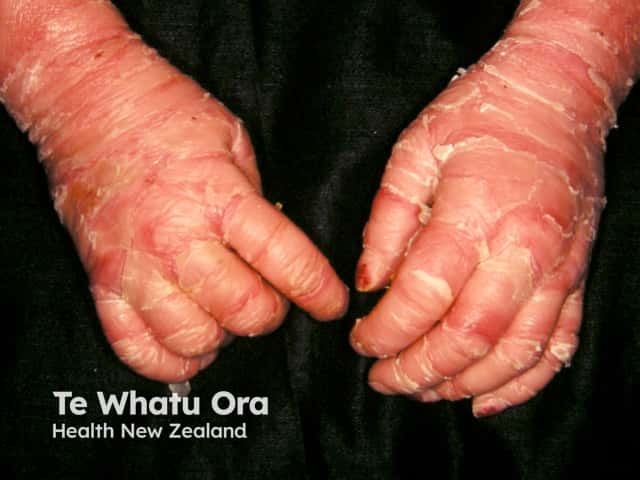
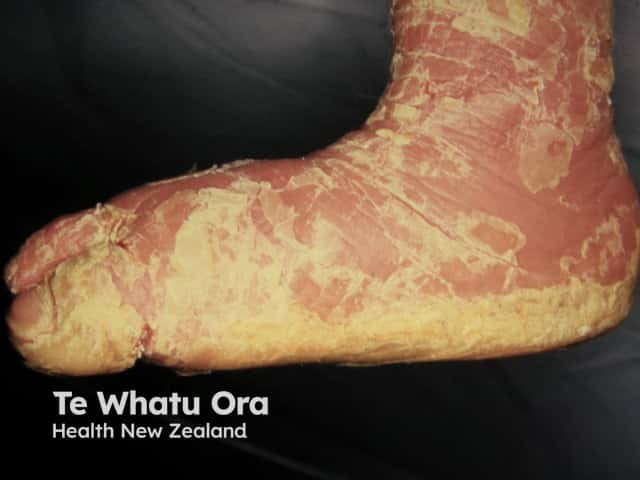
- Improvement with age; blistering is replaced by dark-brown, grey, or white dry or macerated scales and erythema
- Palmoplantar keratoderma.
Common symptoms are anhidrosis, pruritus, fissuring, and decreased joint mobility.
Hair, nails, and mucosal surfaces are usually not involved.
Epidermolytic ichthyosis in an infant
What are the complications of epidermolytic ichthyosis?
Complications of epidermolytic ichthyosis are due to the impaired skin barrier function [2,3].
- Dehydration and electrolyte imbalance due to increased transepidermal water loss.
- Bacterial colonisation of the macerated scales, leading to foul odour, superinfection, and sepsis.
Epidermolytic ichthyosis is associated with atypical naevi [18]. There are also reports of palmoplantar contractures [20].
How is epidermolytic ichthyosis diagnosed?
Given the similarities between various ichthyoses, a diagnosis of epidermolytic ichthyosis can be made using clinical, histopathological, and laboratory findings. The diagnosis can be confirmed with genetic testing [1–3].
- Hyperkeratosis with ‘corrugated cardboard-like’ or ‘cobblestoning’ patterns of scaling are found over flexural or extensor surfaces respectively.
- Histological features of epidermolytic ichthyosis include hyperkeratosis, vacuolar degeneration in cells of the stratum spinosum/granulosum, coarse irregular keratohyaline granules, dyskeratosis, and acantholysis. [see Epidermolytic hyperkeratosis pathology]
- Immunohistochemistry with specific keratin antibodies can be used to identify the type of keratin involved.
- Genetic sequencing can be used to determine the exact site of mutation to predict disease severity and response to treatment.
- Various approaches for prenatal diagnosis have been proposed, including chorionic villus sampling and amniocentesis, fetoscopy with skin biopsy, maternal serum screening, and modern methods such as Raman spectroscopy and optical coherence tomography.
Epidermolytic ichthyosis in an adult
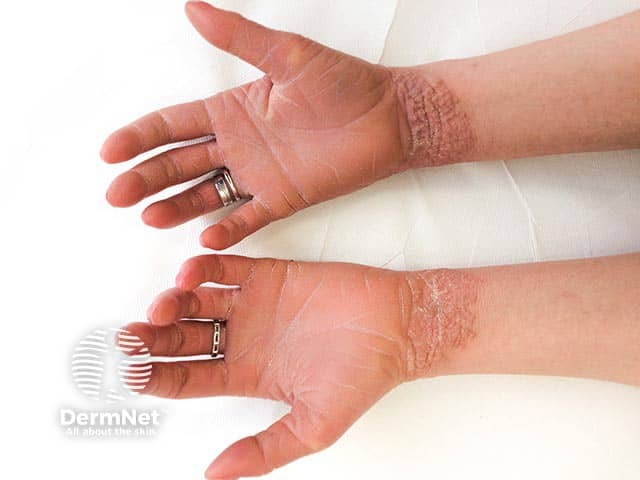
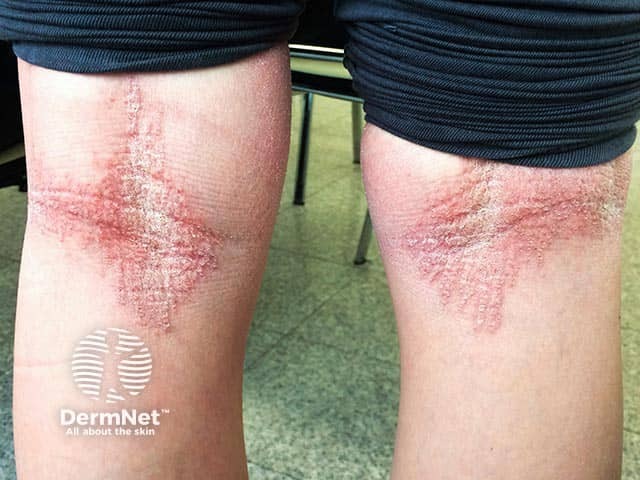
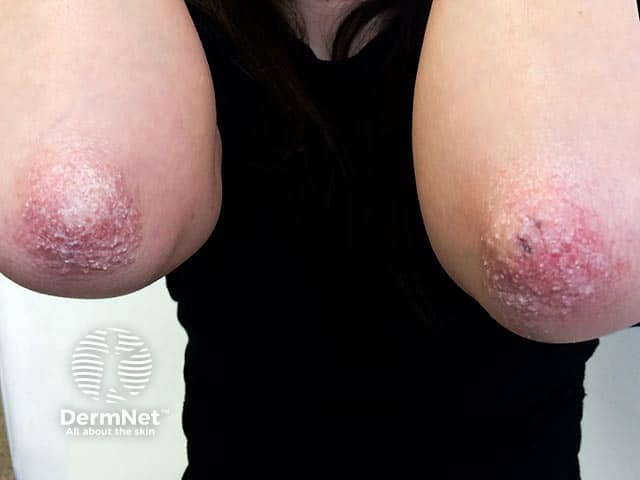
What is the differential diagnosis for epidermolytic ichthyosis?
The differential diagnosis for epidermolytic ichthyosis includes other congenital ichthyoses, vesiculobullous and erosive disorders, and syndromic genodermatoses [2,3].
Other congenital ichthyoses
Other congenital ichthyoses can be a differential diagnosis to epidermolytic ichthyosis include:
- Superficial epidermolytic ichthyosis (ichthyosis bullosa of Siemens) — mutations in keratin gene KRT2
- Lamellar ichthyosis — mutation in transglutaminase gene TGM1
- Ichthyosis vulgaris — mutation in filaggrin gene FLG
- X-linked ichthyosis — mutation in steroid sulfatase gene STS
- Congenital ichthyosiform erythroderma — mutation in TGM1 and lipoxygenase genes ALOX12B, ALOXE3
- Ichthyosis hystrix, Curth–Macklin type — mutation in KRT1
- Ichthyosis with confetti — mutation in KRT10.
Vesicobullous and erosive disorders
Vesicobullous and erosive disorders that could be a differential diagnosis to epidermolytic ichthyosis include:
- Epidermolysis bullosa
- Bullous impetigo
- Staphylococcus scalded skin syndrome
- Herpes simplex
- Congenital erosive and vesicular dermatosis
- Autoimmune blistering disorders (pemphigus, bullous pemphigoid, epidermolysis bullosa acquista, dermatitis herpetiformis, and linear IgA bullous disease).
Syndromic genodermatoses
Syndromic genodermatoses that can be a differential diagnosis to epidermolytic ichthyosis include:
- Netherton syndrome — mutation in SPINK5 gene encoding LEKT1 protein
- Sjögren-Larsson syndrome — mutation in fatty acid metabolism gene FADH
- Conradi-Hünerman syndrome — X-linked mutation in the EBP gene
- CHILD syndrome — mutation in cholesterol gene NSDHL
- KID (keratitis-ichthyosis-deafness) syndrome — mutation in gap junction gene GJB2 encoding connexin 26
- Trichothiodystrophy — mutation in TF2H genes ERCC2, ERCC3, GTF2H5.
What is the treatment for epidermolytic ichthyosis?
There is no cure for epidermolytic ichthyosis, and treatments are limited to management of symptoms depending on the age and sex of the patient, severity of the disease, and location of the skin lesions [2,3]. These include:
- Hydration and lubrication — important to prevent dehydration and electrolyte balance. Glycerol, petrolatum, and other lipid agents are used.
- Antiseptics — antibacterial soaps, chlorhexidine, and sodium hypochlorite (bleach) baths can help prevent infections, which require topical or systemic antibiotics as treatment.
- Keratolytics — the main goal of therapy is to reduce hyperkeratosis. Common keratolytics include α-hydroxy acids, urea, propylene glycol, salicylic acid, N-acetylcysteinamide, liarozole, and calcipotriol.
Retinoids are used to prevent hyperkeratinisation and superinfection in severe cases and have been found to be more effective in patients with KRT10 mutations [21,22]. However, retinoids can paradoxically increase skin fragility and blistering and must be used with caution and careful monitoring [23].
What is the outcome for epidermolytic ichthyosis?
The prognosis of epidermolytic ichthyosis is variable and depends on the severity of the symptoms. There is an immediate risk of dehydration, infection, sepsis, and premature death. Patients who survive experience infection, skin fragility, and blistering episodes throughout their lives, in addition to stress and social isolation accompanying these symptoms [1,2].
Gene therapy has been demonstrated to be effective in improving tonofilament stability in KRT10 mutant primary keratinocytes [24] and remains the best hope for these patients.
References
- Rout PD, Nair A, Gupta A, Kumar P. Epidermolytic hyperkeratosis: clinical update. Clin Cosmet Investig Dermatol 2019; 12: 333–44. DOI: 10.2147/ccid.S166849. PubMed Central
- Rice AS, Crane JS. Epidermolytic Hyperkeratosis (Bullous Ichthyosiform Erythroderma). Statpearls. Treasure Island (FL): StatPearls Publishing, 2020. PubMed
- Lacz NL, Schwartz RA, Kihiczak G. Epidermolytic Hyperkeratosis: A keratin 1 or 10 mutational event. Int J Dermatol 2005; 44: 1–6. DOI: 10.1111/j.1365-4632.2004.02364.x. PubMed
- Chipev CC, Korge BP, Markova N, Bale SJ, et al. A leucine proline mutation in the H1 subdomain of keratin 1 causes epidermolytic hyperkeratosis. Cell 1992; 70: 821–8. DOI: 10.1016/0092-8674(92)90315-4. PubMed
- Rothnagel JA, Dominey AM, Dempsey LD, Longley MA, et al. Mutations in the rod domains of keratins 1 and 10 in epidermolytic hyperkeratosis. Science. 1992; 257: 1128–30. DOI: 10.1126/science.257.5073.1128. PubMed
- Cheng J, Syder AJ, Yu QC, Letai A, et al. The genetic basis of epidermolytic hyperkeratosis: a disorder of differentiation-specific epidermal keratin genes. Cell 1992; 70: 811–9. DOI: 10.1016/0092-8674(92)90314-3. PubMed
- Fuchs E, Esteves RA, Coulombe PA. Transgenic mice expressing a mutant keratin 10 gene reveal the likely genetic basis for epidermolytic hyperkeratosis. Proc Natl Acad Sci USA 1992; 89: 6906–10. DOI: 10.1073/pnas.89.15.6906. PubMed Central
- Reis A, Hennies HC, Langbein L, Digweed M, et al. Keratin 9 gene mutations in epidermolytic palmoplantar keratoderma (Eppk). Nat Genet 1994; 6: 174–9. DOI: 10.1038/ng0294-174. PubMed
- Wang P, Kang XJ, Tang XH, Liu JY, et al. Six generations of epidermolytic palmoplantar keratoderma, associated with a Krt9 R163w mutation. Cancer Genet 2016; 209: 515–24. DOI: 10.1016/j.cancergen.2016.10.002. PubMed
- Fuchs E, Green H. The expression of keratin genes in epidermis and cultured epidermal cells. Cell 1978; 15: 887–97. DOI: 10.1016/0092-8674(78)90273-8. PubMed
- Schweizer J, Kinjo M, Fürstenberger G, Winter H. Sequential Expression of Mrna-Encoded Keratin Sets in Neonatal Mouse Epidermis: Basal Cells with Properties of Terminally Differentiating Cells. Cell 1984; 37: 159–170. DOI: 10.1016/0092-8674(84)90311-8. Journal
- Woodcock-Mitchell J, Eichner R, Nelson WG, Sun TT. Immunolocalization of keratin polypeptides in human epidermis using monoclonal antibodies. J Cell Biol 1982; 95: 580–8. DOI: 10.1083/jcb.95.2.580. PubMed
- Chen PJ, Li CX, Wen J, Peng YS, et al. S159p mutation of keratin 10 gene causes severe form of epidermolytic hyperkeratosis. J Eur Acad Dermatol Venereol 2016; 30: e102–4. DOI: 10.1111/jdv.13345. PubMed
- Syder AJ, Yu QC, Paller AS, Giudice G, et al. Genetic mutations in the K1 and K10 genes of patients with epidermolytic hyperkeratosis. Correlation between location and disease severity. J Clin Invest 1994; 93: 1533–42. DOI: 10.1172/jci117132. PubMed Central
- Terheyden P, Grimberg G, Hausser I, Rose C, et al. Recessive epidermolytic hyperkeratosis caused by a previously unreported termination codon mutation in the keratin 10 gene. J Invest Dermatol 2009; 129: 2721–23. DOI: 10.1038/jid.2009.131. PubMed
- Tsubota A, Akiyama M, Kanitakis J, Sakai K, et al. Mild recessive bullous congenital ichthyosiform erythroderma due to a previously unidentified homozygous keratin 10 nonsense mutation. J Invest Dermatol 2008; 128: 1648–52. DOI: 10.1038/sj.jid.5701257. PubMed
- Paller AS, Syder AJ, Chan YM, Yu QC, et al. Genetic and clinical mosaicism in a type of epidermal nevus. N Engl J Med 1994; 331: 1408–15. DOI: 10.1056/nejm199411243312103. PubMed
- Conlin PA, Rapini RP. Epidermolytic hyperkeratosis associated with melanocytic nevi: a report of 53 cases. Am J Dermatopathol 2002; 24: 23–5. DOI: 10.1097/00000372-200202000-00004. PubMed
- Irvine AD, McLean WH. Human keratin diseases: the increasing spectrum of disease and subtlety of the phenotype-genotype correlation. Br J Dermatol 1999; 140: 815–28. DOI: 10.1046/j.1365-2133.1999.02810.x. PubMed
- Palmer AK, Louis DS. Flexion contractures of the hand associated with ichthyosis hystrix. Case report. J Bone Joint Surg Am 1976; 58: 143–4. PubMed
- Li H, Törmä H. Retinoids reduce formation of keratin aggregates in heat-stressed immortalized keratinocytes from an epidermolytic ichthyosis patient with a Krt10 mutation*. Acta Derm Venereol 2013; 93: 44–9. DOI: 10.2340/00015555-1368. PubMed
- Virtanen M, Gedde-Dahl T, Jr., Mörk NJ, Leigh I, et al. Phenotypic/genotypic correlations in patients with epidermolytic hyperkeratosis and the effects of retinoid therapy on keratin expression. Acta Derm Venereol 2001; 81: 163–70. DOI: 10.1080/000155501750376221. PubMed
- Lacour M, Mehta-Nikhar B, Atherton DJ, Harper JI. An appraisal of acitretin therapy in children with inherited disorders of keratinization. Br J Dermatol 1996; 134: 1023–9. PubMed
- March OP, Lettner T, Klausegger A, Ablinger M, et al. Gene editing-mediated disruption of epidermolytic ichthyosis-associated Krt10 alleles restores filament stability in keratinocytes. J Invest Dermatol 2019; 139: 1699–1710.e1696. DOI: 10.1016/j.jid.2019.03.1146. PubMed
On DermNet
- Epidermolytic hyperkeratosis pathology
- Ichthyosis
- Ichthyosis vulgaris
- Ichthyosis prematurity syndrome
- Recessive X-linked ichthyosis
- Netherton syndrome
- Epidermal naevus
Other websites
- Epidermolytic ichthyosis — NIH Genetic and Rare Diseases Information Center
- Epidermolytic Hyperkeratosis — OMIM
- Epidermolytic ichthyosis — First Skin Foundation
- Epidermolytic ichthyosis — National Organization for Rare Disorders (NORD)