
Sturge–Weber syndrome — extra information
Introduction Demographics Causes Clinical features Complications Diagnosis Differential diagnoses Treatment Outcome
What is Sturge–Weber syndrome?
Sturge–Weber syndrome is a rare, congenital, and non-inherited neurocutaneous disorder characterised by capillary malformation on the facial skin (port-wine stain) and capillary-venous malformations in the brain and in the eyes [1].
Who gets Sturge–Weber syndrome?
It is estimated that approximately one in every 20,000 to 50,000 babies are born with Sturge–Weber syndrome [2]. As it is caused by a somatic mosaic mutation, it is not inherited from the parents.
What causes Sturge–Weber syndrome?
Sturge–Weber syndrome is caused by a somatic mosaic mutation of the GNAQ gene on chromosome 9 [3]. This means that the mutation in the gene has occurred in the body cells after the formation of the zygote. GNAQ regulates intracellular signalling pathways. The mutation gives rise to uncontrolled formation or maturation of capillaries in affected cells.
What are the clinical features of Sturge–Weber syndrome?
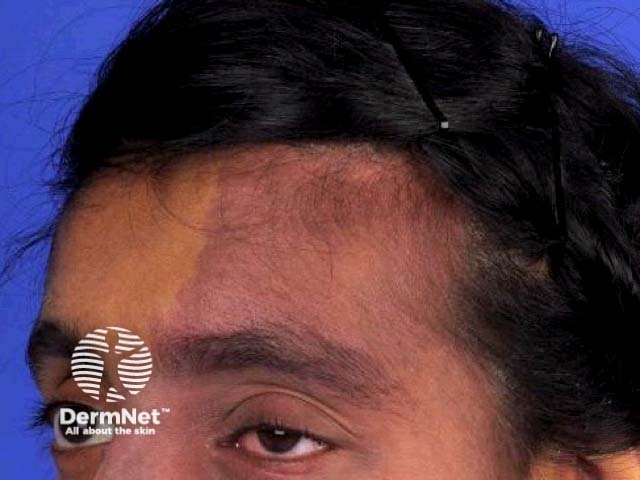
Sturge–Weber syndrome is characterised by vascular malformations on the face and in the eye and brain of affected individuals. These are present at birth.
- Port-wine stains are the most common type of vascular malformation, affecting approximately three in 1000 infants, but most are not associated with Sturge–Weber syndrome [4].
- Port-wine stains in Sturge–Weber syndrome are typically in the distribution of the first and second division of the trigeminal nerve on the forehead and upper eyelid [5]. They may also affect both sides of the face [6].
- Leptomeningeal vascular malformations arise inside the brain on the same side as the port-wine stain.
- Leptomeningeal vascular malformations may also occur without a port-wine stain [7].
Neurological and ophthalmological signs in Sturge-Weber syndrome are progressive and usually develop in the first two years of life. These can include:
- Seizures and epilepsy
- Hemiparesis and stroke-like events
- Behavioural problems
- Visual field defects and glaucoma
- Growth hormone deficiency.
What are the complications of Sturge–Weber syndrome?
The complications of Sturge–Weber syndrome depend on the extent of vascular malformations and other clinical features. They are extremely variable.
How is Sturge–Weber syndrome diagnosed?
The diagnosis of Sturge–Weber syndrome is based on finding the characteristic trigeminal port-wine stains and leptomeningeal capillary-venous malformations. A diagnosis based on leptomeningeal lesions alone depends on the development of symptoms.
If there are neurological symptoms or findings, magnetic resonance imaging (MRI) of the brain is undertaken with gadolinium contrast to detect leptomeningeal capillary-venous malformations.
What is the differential diagnosis for Sturge–Weber syndrome?
The characteristics of other vascular malformation syndromes are described below.
- Klippel–Trénaunay syndrome has more extensive capillary malformations, involves the limbs and trunk, and is associated with hypertrophy of the affected limb.
- Parkes–Weber syndrome presents with a large capillary malformation on an extremity and hypertrophy of the affected limb. There are multiple, fast-flowing arteriovenous shunts.
- Servelle–Martorell syndrome, a rare congenital angiodysplastic disease, is associated with progressive limb hypotrophy.
- Proteus syndrome presents with asymmetrical and disproportionate overgrowth of body parts. The syndrome results from a somatic activating mutation in the AKT1 oncogene.
- CLOVES syndrome presents with congenital lipomatous overgrowth, vascular malformation, epidermal naevus, spinal/skeletal anomalies/scoliosis. The syndrome results from somatic mosaic activating mutations in the PIK3CA gene.
What is the treatment for Sturge–Weber syndrome?
There is no specific treatment for Sturge–Weber syndrome. Treatment consists of managing the cutaneous, neurological and ocular symptoms, with limited success.
Treatment of seizures in patients with Sturge–Weber syndrome with antiepileptics is not always successful [8]. No single treatment appears to be superior to others.
Port-wine stains can be treated with pulsed-dye laser with variable results [9]. Due to the generally poor results of pulsed-dye lasers alone, topical antiangiogenic agents are being trialled as adjunctive therapies.
As glaucoma is a common complication of Sturge-Weber syndrome, regular eye checks with an ophthalmologist are recommended. Glaucoma is treated surgically and medically [10].
Infants diagnosed with Sturge–Weber syndrome should be treated with low-dose aspirin [11,12]. The antithrombotic therapy may prevent the progression of the disease, which can impair blood flow to the brain resulting in neuronal damage.
What is the outcome for Sturge–Weber syndrome?
The prognosis of Sturge–Weber syndrome depends on the extent of involvement of the brain and the skin. Extensive port-wine stains are associated with a higher risk of epilepsy and glaucoma, while bilateral leptomeningeal vascular malformations are associated with learning and intellectual disability [13]. The onset of seizures before the age of one has a significant effect on cognitive and motor function in children with Sturge–Weber syndrome [14].
It is estimated that approximately one in two adults with Sturge–Weber syndrome have neurological defects, even in those who were initially asymptomatic.
References
- Enjolras O, Riche MC, Merland JJ. Facial port-wine stains and Sturge–Weber syndrome. Pediatrics 1985; 76: 48–51. PubMed
- Comi AM. Update on Sturge-Weber syndrome: diagnosis, treatment, quantitative measures, and controversies. Lymphat Res Biol 2007; 5: 257–64. DOI: 10.1089/lrb.2007.1016. PubMed
- Shirley MD, Tang H, Gallione CJ, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med 2013; 368: 1971–9. DOI: 10.1056/NEJMoa1213507. PubMed
- Piram M, Lorette G, Sirinelli D, Herbreteau D, Giraudeau B, Maruani A. Sturge-Weber syndrome in patients with facial port-wine stain. Pediatr Dermatol 2012; 29: 32–7. DOI: 10.1111/j.1525-1470.2011.01485.x. PubMed
- Bodensteiner JB. Sturge-Weber syndrome. Facial Plast Surg Clin North Am 2001; 9: 569–76. PubMed
- Tallman B, Tan OT, Trainor S, et al. Location of port-wine stains and the likelihood of ophthalmic and/or central nervous system complications. Pediatrics 1991; 87: 323–7. PubMed
- Aydin A, Cakmakçi H, Kovanlikaya A, Dirik E. Sturge-Weber syndrome without facial nevus. Pediatr Neurol 2000; 22: 400–2. DOI: 10.1016/s0887-8994(00)00127-2. Journal
- Arzimanoglou A, Aicardi J. The epilepsy of Sturge-Weber syndrome: clinical features and treatment in 23 patients. Acta Neurol Scand Suppl 1992; 140: 18–22. DOI: 10.1111/j.1600-0404.1992.tb04465.x. PubMed
- Renfro L, Geronemus RG. Anatomical differences of port-wine stains in response to treatment with the pulsed dye laser. Arch Dermatol 1993; 129: 182–8. PubMed
- Javaid U, Ali MH, Jamal S, Butt NH. Pathophysiology, diagnosis, and management of glaucoma associated with Sturge-Weber syndrome. Int Ophthalmol 2018; 38: 409–16. DOI: 10.1007/s10792-016-0412-3. PubMed
- Lance EI, Sreenivasan AK, Zabel TA, Kossoff EH, Comi AM. Aspirin use in Sturge-Weber syndrome: side effects and clinical outcomes. J Child Neurol 2013; 28: 213–18. DOI: 10.1177/0883073812463607. PubMed
- Bay MJ, Kossoff EH, Lehmann CU, Zabel TA, Comi AM. Survey of aspirin use in Sturge-Weber syndrome. J Child Neurol 2011; 26: 692–702. DOI: 10.1177/0883073810388646. PubMed
- Day AM, McCulloch CE, Hammill AM, et al. Physical and family history variables associated with neurological and cognitive development in Sturge-Weber syndrome. Pediatr Neurol 2019; 96: 30–6. DOI: 10.1016/j.pediatrneurol.2018.12.002. PubMed
- Luat AF, Behen ME, Chugani HT, Juhász C. Cognitive and motor outcomes in children with unilateral Sturge-Weber syndrome: effect of age at seizure onset and side of brain involvement. Epilepsy Behav 2018; 80: 202–7. DOI: 10.1016/j.yebeh.2018.01.012. PubMed
On DermNet
- Capillary vascular malformations
- Proteus syndrome
- CLOVES syndrome
- Facial red vein and vascular birthmark treatments
Other websites
- Sturge–Weber Syndrome — Healthline
- Sturge–Weber Syndrome — Medscape
- The Sturge–Weber Foundation