
Lanadelumab — extra information
Introduction Introduction How it works Indication How to use Contraindications Warnings and precautions Dosage modification Use in specific populations Potential drug interactions Possible side effects Immunogenicity
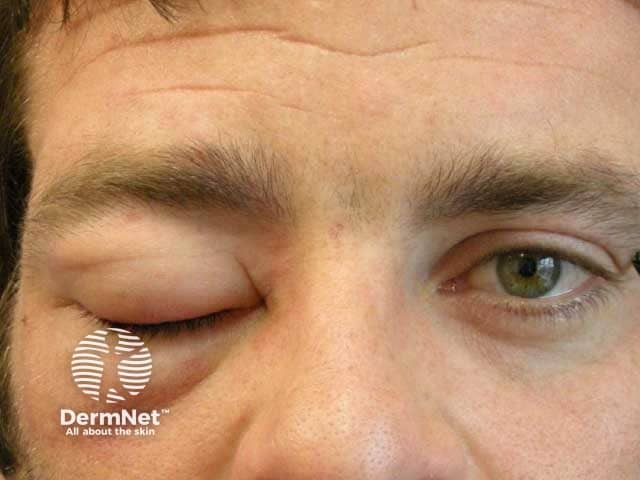
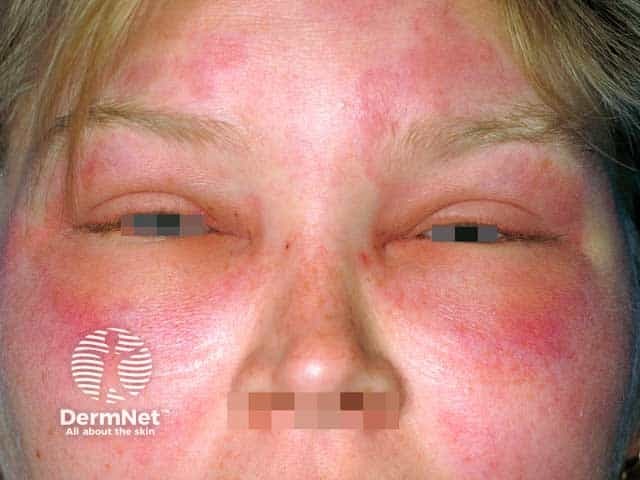
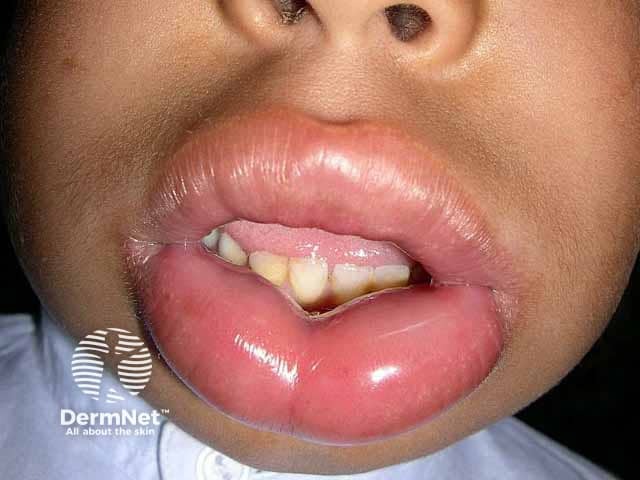
What is hereditary angioedema?
Hereditary angioedema is a rare, autosomal dominant disease that is associated with a deficiency or dysfunction of C1-esterase-inhibitor (C1-INH) activity. Hereditary angioedema causes spontaneous, recurrent, and potentially life-threatening swelling in various regions of the body, including the larynx.
What is lanadelumab?
Lanadelumab (also called lanadelumab-flyo) is a prescription biological treatment used in patients aged 12 years and over to prevent severe swelling attacks due to hereditary angioedema. It is the first monoclonal antibody available for hereditary angioedema.
Lanadelumab was approved by the US Food and Drug Administration (FDA) in August 2018, in Canada in September 2018, and by the European Commission in November 2018. Regulatory applications for marketing lanadelumab are also being considered in Australia. Lanadelumab is not currently available in New Zealand.
Lanadelumab is marketed under the brand Takhzyro™ by Dyax Corp (a subsidiary of Shire) based in Burlington, MA, USA.
How does lanadelumab work?
Lanadelumab is a fully human immunoglobulin G1, kappa light chain monoclonal antibody that provides targeted inhibition of plasma kallikrein, a critical regulator of hereditary angioedema attacks.
- The activity of plasma kallikrein is regulated by the activity of C1-INH.
- C1-INH deficiency or dysfunction leads to uncontrolled increases in plasma kallikrein activity.
- Plasma kallikrein is a protease that works by splitting the high molecular weight kininogen (a coagulation protein) to generate the pro-inflammatory peptide bradykinin. Bradykinin is a potent vasodilator and increases vascular permeability, which is the cause of the swelling and pain associated with hereditary angioedema.
- Lanadelumab obstructs the proteolytic active site of plasma kallikrein, preventing the splitting of kininogen into bradykinin.
- Lanadelumab, therefore, decreases plasma kallikrein and bradykinin activity in patients with hereditary angioedema.
What is the indication for lanadelumab?
Lanadelumab is indicated to prevent recurrent attacks of hereditary angioedema in patients aged 12 years and older.
- It is not intended for the treatment of acute hereditary angioedema attacks.
- Treatment should be started under the supervision of a physician experienced in the management of patients with hereditary angioedema.
How is lanadelumab administered?
Lanadelumab may be self-administered by the patient or administered by a caregiver after undergoing training on subcutaneous injection technique by a healthcare professional.
- The recommended starting dose is 300 mg lanadelumab by subcutaneous injection every 2 weeks.
- The injection should be restricted to the recommended injection sites — the abdomen, the thighs, and the upper outer arms — with the injection site being rotated as required.
- In patients who are free of swelling attacks while on treatment, the dose may be reduced to 300 mg every 4 weeks.
- If a dose of lanadelumab is missed, the missed dose should be administered as soon as possible, while ensuring at least 10 days between doses.
Contraindications to lanadelumab
The main contraindication to lanadelumab is hypersensitivity to the active substance or to any of the excipients.
Warnings and precautions
Traceability
To improve the traceability of biological medicinal products, the name and the batch number of the administered lanadelumab should be clearly recorded.
Hypersensitivity reactions
Hypersensitivity reactions to lanadelumab have been observed. In case of a severe hypersensitivity reaction, the administration of lanadelumab must be stopped immediately and appropriate treatment initiated.
General
Lanadelumab is not intended for the treatment of acute attacks of hereditary angioedema. In the case of breakthrough angioedema, individualised treatment should be initiated with approved rescue medication.
There are no available clinical data on the use of lanadelumab in hereditary angioedema patients with normal C1-INH activity.
Dosage modification
Patients with renal impairment
No studies have been conducted in patients taking lanadelumab who have severe renal impairment. Renal impairment is not expected to affect the exposure to lanadelumab or its safety profile. No dose adjustment is required in patients with renal impairment.
Patients with hepatic impairment
No dose adjustment is required in patients taking lanadelumab who have hepatic impairment. Hepatic impairment is not expected to affect exposure to lanadelumab. There are no studies in patients with moderate or severe hepatic impairment.
Older people
No dose adjustment is required for patients above 65 years of age because age is not expected to affect the exposure to lanadelumab.
Children
The safety and efficacy of lanadelumab in children aged less than 12 years have not been established. No data are available.
Use of lanadelumab in specific populations
Pregnant women
There are no available data on the use of lanadelumab in pregnant women to inform any drug-associated risks. Monoclonal antibodies such as lanadelumab can cross the placenta during the third trimester of pregnancy with potentially harmful effects on the fetus. Studies of lanadelumab in pregnant cynomolgus monkeys, treated with 33 times the maximum recommended dose for humans, revealed no evidence of harm to the developing fetus.
Nursing mothers
It is not known whether lanadelumab or its metabolites are excreted in human milk. Because many drugs are excreted in human milk, mothers should discontinue nursing prior to using lanadelumab. Available pharmacokinetic data in cynomolgus monkeys have shown excretion of lanadelumab in milk at approximately 0.2% of the maternal plasma level.
Children
The safety and efficacy of lanadelumab in children under 12 years of age have not been established.
Older people
In clinical trials, no overall differences in safety or effectiveness have been observed between patients over 65 years of age and younger patients treated with lanadelumab.
Individuals of fertile age
The effect of lanadelumab on fertility has not been evaluated in humans. Lanadelumab had no effect on male or female fertility in cynomolgus monkeys.
What are potential drug interactions with lanadelumab?
No dedicated drug–drug interaction studies have been conducted with lanadelumab. Based on its characteristics, no pharmacokinetic interactions with co-administered medicinal products are expected.
Interaction with aPPT assay
- Lanadelumab can increase the activated partial thromboplastin time (aPTT) due to an interaction with the aPTT assay.
- Reagents used in the aPTT laboratory test initiate intrinsic coagulation through the activation of plasma kallikrein in the contact activation system.
- Inhibition of plasma kallikrein by lanadelumab can increase aPTT in this assay.
What are the possible side effects of lanadelumab?
The most common side effects in patients receiving lanadelumab in clinical trials are described below.
Side effects with over a 10% incidence
At a dose of lanadelumab 300 mg every 2 weeks, the following adverse reactions where seen:
- Injection site reactions, including pain, erythema, and bruising (56%)
- Upper respiratory tract infection (44%)
- Headache (33%)
- Myalgia (11%).
At a dose of lanadelumab 300 mg every 4 weeks, the following adverse reactions where seen:
- Injection site reactions (45%)
- Upper respiratory tract infection (31%)
- Headache (21%).
Side effects of 1–10% incidence
Adverse reactions of 1–10% in incidence seen with lanadelumab include:
- Increased alanine transferase (ALT) and aspartate transferase (AST) liver enzymes (2%)
- Hypersensitivity (1%) — symptoms of a serious allergic reaction include urticaria, itching/swelling (especially of the face, tongue, and throat), severe dizziness, and difficulty breathing.
At a dose of lanadelumab 300 mg every 2 weeks, the following adverse reactions were seen:
- Rash (4%)
- Dizziness (4%)
- Diarrhoea (4%).
At a dose of lanadelumab 300 mg every 4 weeks, the following adverse reactions where seen:
- Rash (10%)
- Dizziness (10%).
Immunogenicity
Treatment with lanadelumab has been associated with the development of anti-drug antibodies in 10 out of 84 patients (11.9%). All antibody titres were low. The anti-drug antibody response was transient in 20% of subjects positive for anti-drug antibodies; two out of 84 lanadelumab-treated patients (2.4%) tested positive for neutralising antibodies.
The development of anti-drug antibodies, including neutralising antibodies against lanadelumab, did not appear to adversely affect the pharmacokinetic and pharmacodynamic profiles or the clinical response.
Approved datasheets are the official source of information for medicines, including approved uses, doses, and safety information. Check the individual datasheet in your country for information about medicines.
We suggest you refer to your national drug approval agency such as the Australian Therapeutic Goods Administration (TGA), US Food and Drug Administration (FDA), UK Medicines and Healthcare products regulatory agency (MHRA) / emc, and NZ Medsafe, or a national or state-approved formulary eg, the New Zealand Formulary (NZF) and New Zealand Formulary for Children (NZFC) and the British National Formulary (BNF) and [British National Formulary for Children ](http://bnfc.nice.org.uk/ (BNFC).
References
- Riedl MA, Bernstein JA, Craig T, et al. An open-label study to evaluate the long-term safety and efficacy of lanadelumab for prevention of attacks in hereditary angioedema: design of the HELP study extension. Clin Transl Allergy 2017; 7: 36. DOI: 10.1186/s13601-017-0172-9. PubMed
- Banerji A, Busse P, Shennak M, et al. Inhibiting plasma kallikrein for hereditary angioedema prophylaxis. N Engl J Med 2017; 376: 717–28. PubMed
- Banerji A, Riedl MA, Bernstein JA, et al. Effect of lanadelumab compared with placebo on prevention of hereditary angioedema attacks: a randomized clinical trial. JAMA 2018; 320: 2108–21. DOI: 10.1001/jama.2018.16773. PubMed
- Zuraw BL, Banerji A, Bernstein JA, et al. US Hereditary Angioedema Association Medical Advisory Board 2013 recommendations for the management of hereditary angioedema due to C1 inhibitor deficiency. J Allergy Clin Immunol Pract 2013; 1: 458–67. DOI: 10.1016/j.jaip.2013.07.002. PubMed
- Cicardi M, Aberer W, Banerji A, et al. Classification, diagnosis, and approach to treatment for angioedema: consensus report from the Hereditary Angioedema International Working Group. Allergy 2014; 69: 602–16. DOI: 10.1111/all.12380. PubMed
On DermNet
- Key clinical-trial evidence for lanadelumab
- Hereditary angioedema
- Angioedema
- Injected skin treatments
- Biological treatments
Other websites
- Lanadelumab — US Food and Drug Administration prescribing information (pdf)
- Lanadelumab — Medscape data sheet
- Lanadelumab — Product monograph Canada (pdf)