
Alpha-1-antitrypsin deficiency panniculitis — extra information
Introduction - alpha-1-antitrypsin deficiency disease Introduction - panniculitis Demographics Causes Clinical features Diagnosis Treatment General measures Oral medications Outcome
What is alpha-1-antitrypsin deficiency disease?
Alpha-1-antitrypsin is a serine protease inhibitor. Alpha-1-antitrypsin deficiency disease is an inherited metabolic disorder in which this protein is absent or defective.
Alpha-1-antitrypsin deficiency disease most commonly presents as lung disease (emphysema) or gastrointestinal disease (liver cirrhosis). It can also rarely affect the skin.
Alpha-1-antitrypsin deficiency is abbreviated as A1AT deficiency.
What is panniculitis?
Panniculitis is an inflammation of the subcutaneous adipose tissue (the fat underneath the skin).
Panniculitis due to alpha-1-antitrypsin deficiency is rare.
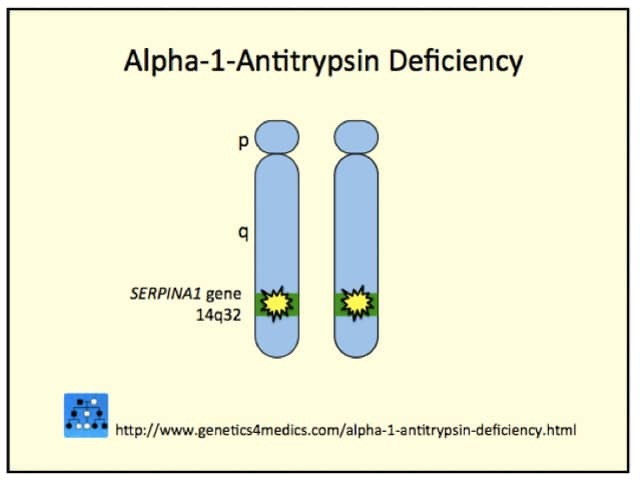
*Image courtesy Genetics 4 Medics
Who gets alpha-1-antitrypsin deficiency panniculitis?
Alpha-1-antitrypsin deficiency is most prevalent in Caucasians of European and North American descent.
Males and females of any age are equally affected by the panniculitis associated with alpha-1-antitrypsin deficiency, and it can rarely arise in children.
As alpha-1-antitrypsin deficiency is under-diagnosed, the true prevalence of panniculitis due to alpha-1-antitrypsin deficiency is difficult to ascertain. One study reported panniculitis affected 0.9% of ZZ homozygotes for alpha-1-antitrypsin deficiency.
Panniculitis may be preceded by trauma. This is because an injury causes increased protease activation and a cascade of inflammatory events intended to heal the wound.
What causes alpha-1-antitrypsin deficiency?
Genetics
Alpha-1-antitrypsin deficiency is usually due to a mutation on chromosome 14.
Alpha-1-antitrypsin deficiency is an autosomal co-dominant disorder, meaning that two different versions (alleles) of a gene are expressed in an individual. Each allele makes a slightly different protein. Both alleles influence the characteristics and severity of the deficiency disease.
There are over 90 known mutant alleles of the alpha-1-antitrypsin gene, and they are classified based on their acid starch gel mobility (F=fast, M=medium, S = slow, Z = very slow). A normal level of a variant enzyme with altered activity can also cause clinical disease.
The genotype (a person’s genetic makeup) and their combination of these genes determine the degree of the deficiency. The normal genotype is ‘MM’ has a normal level of alpha-1-antitrypsin. Patients with two ‘ZZ’ genes (homozygotes) are 85% deficient in alpha-1-antitrypsin. This type is the most commonly associated with emphysema.
Pathogenesis
Alpha-1-antitrypsin is mostly produced in the liver and regulates several proteolytic enzymes including trypsin, elastase, chymotrypsin, factor VIII, collagenase and kallikrein. Lack of alpha-1-antitrypsin leads to increased activity of these enzymes and localised tissue destruction.
What are the clinical features of alpha-1-antitrypsin deficiency?
Deficiency of alpha-1-antitrypsin leads to inflammation and necrosis in various organs.
Lung disease
- Alpha-1-antitrypsin usually protects the lungs from elastase enzymes produced by neutrophils.
- When alpha-1-antitrypsin is deficient, elastase enzymes can damage and scar lung tissue causing emphysema.
- Asthma, bronchiectasis and chronic obstructive pulmonary disease are also more prevalent.
- Lung disease is much more severe in smokers than in non-smokers with alpha-1-antitrypsin deficiency.
Liver disease
- Abnormal alpha-1-antitrypsin protein accumulates within the liver cells (hepatocytes) and can cause hepatitis.
- Hepatitis can present with a swollen abdomen, leg oedema or jaundice.
- Liver disease is accelerated by drinking alcohol.
- Later, cirrhosis (scarring of the liver) occurs.
- This increases the risk of hepatocellular carcinoma.
Pancreatic disease
- Elastase is a pancreatic enzyme.
- Increased elastase due to alpha-1-antitrypsin deficiency can cause chronic pancreatitis.
- Chronic pancreatitis leads to steatorrhoea, malabsorption, vitamin deficiency (A, D, E, K, B12), diabetes mellitus, and weight loss.
Kidney disease
- Membranoproliferative glomerulonephritis
Panniculitis
In alpha-1-antitrypsin deficiency disease:
- Panniculitis is due to unopposed enzyme action and protein breakdown within the skin
- Initially erythematous plaques and nodules develop on the trunk or proximal limbs
- Nodules may be widespread over the legs, arms, trunk, or face
- The nodules may ulcerate, bleed, and leak oily material
- The patient may be febrile.
The panniculitis associated with alpha-1-antitrypsin deficiency disease does not improve with antibiotics or systemic steroids.
How are alpha-1-antitrypsin deficiency and associated panniculitis diagnosed?
Diagnosis of alpha-1-antitrypsin deficiency
Alpha-1-antitrypsin deficiency disease may be suspected by the clinical features, especially emphysema, chronic hepatitis, and liquefying panniculitis. Several tests may be arranged to confirm the diagnosis.
Serum alpha-1-antitrypsin levels
- Serum alpha-1-antitrypsin levels can confirm if a patient is deficient or not.
- Note that alpha-1-antitrypsin can be misleadingly raised during an inflammatory state, as it is an acute phase protein.
Alpha-1-antitrypsin phenotype and genotyping
- If the disease is highly suspected and alpha-1-antitrypsin levels are normal, an iso-electrophoretic mobility study (alpha-1-antitrypsin phenotype) can be useful.
- Genotyping can detect abnormal alleles in an individual.
Respiratory investigations
- Pulmonary function tests can help to establish if there is emphysema
- Chest X-ray may show characteristic lung changes (emphysema at the lung bases)
- High-Resolution CT (HRCT) scanning can help to identify basal emphysema and bronchiectasis
Diagnosis of panniculitis associated with alpha-1-antitrypsin deficiency
Blood tests
- Inflammatory markers (erythrocyte sedimentation rate and C-reactive protein) may be elevated.
- Antinuclear antibodies and rheumatoid factor may be present in low titre.
- Chronic disease can cause normocytic anaemia and hypoalbuminaemia.
Skin biopsy
A deep incisional skin biopsy is required for panniculitis to include deep adipose tissue. Specific characteristics of alpha-1-antitrypsin deficiency panniculitis pathology include:
- Predominantly lobular inflammation, with some septal involvement and extensive tissue destruction.
- In early disease, neutrophils infiltrate between collagen bundles in the reticular dermis.
- The collagen in the dermis is gradually dissolved, and the epidermis can liquefy.
- The splaying of the neutrophils may progress to involve fibrous septa and lobules of the subcutaneous fat.
- In later stages, histiocytes and lipophages are seen, sometimes with giant cell accumulation.
- Leucocytoclastic and lymphocytic vasculitis can arise in areas of dense inflammation as well as phlebothrombosis and haemorrhage.
- Skip areas of normal fat next to necrotic areas of fat can be a feature.
- Areas heal with fibrosis and sometimes, dystrophic calcification.
- Direct immunofluorescence may detect intravascular deposits of complement C3 and IgM in the dermis of uncertain significance.
What is the treatment for alpha-1-antitrypsin deficiency and its associated panniculitis?
General measures
- Avoid trauma and surgery to the area if possible
- It is essential to avoid smoking and alcohol.
- Avoid occupational exposures that may increase the risk of airways disease (eg, mining, construction work)
- Annual influenza vaccination is recommended
Oral medications
- Tetracycline antibiotics (doxycycline, minocycline) can be effective for mild panniculitis. It is thought the anticollagenase activity of tetracyclines can help re-establish the balance between protease and antiprotease enzymes.
- Dapsone and colchicine can reduce the inflammation associated with panniculitis.
Systemic corticosteroids, antimalarials, immunosuppressants and danazol are ineffective in alpha-1-antitrypsin deficiency disease.
Enzyme replacement therapy
Enzyme replacement therapy with exogenous alpha-1-antitrypsin concentrate (Prolastin®) infused intravenously has been reported to be effective. The initial recommended dose of 60 mg/kg once weekly may be adjusted according to response. The dose required and duration of treatment varies according to case reports. Its use may be limited by high cost and limited availability.
Other measures
- Plasma exchange
- Genetically engineered alpha-1-antitrypsin
- Organ transplantation (lungs/liver) for patients with advanced disease
What is the outcome for alpha-1-antitrypsin deficiency and its associated panniculitis?
The prognosis and life expectancy of a patient with alpha-1-antitrypsin deficiency depend on phenotype, the level of enzyme deficiency, and the associated hepatic and pulmonary damage.
Complete resolution of panniculitis associated with alpha-1-antitrypsin deficiency has been described within four weeks of treatment with enzyme replacement therapy. It may recur, in which case indefinite treatment may be required.
Chronic ulceration can complicate the associated panniculitis, and when areas do finally heal they may form atrophic scarring.
References
- Köhnlein T, Welte T. Alpha-1 antitrypsin deficiency: pathogenesis, clinical presentation, diagnosis, and treatment. The American Journal of Medicine. 2008 Jan 31;121(1):3-9. PubMed
- Requena L, Yus ES. Panniculitis. part II. mostly lobular panniculitis. Journal of the American Academy of Dermatology. 2001 Sep 30;45(3):325-64. PubMed
- Stone H, Pye A, Stockley RA. Disease associations in alpha-1-antitrypsin deficiency. Respiratory Medicine. 2014 Feb 28;108(2):338-43. PubMed
- Su WD, Smith KC, Pittelkow MR, Winkelmann RK. [alpha] 1-Antitrypsin Deficiency Panniculitis: A Histopathologic and Immunopathologic Study of Four Cases. The American Journal of Dermatopathology. 1987 Dec 1;9(6):483-90. PubMed
- Smith KC, Su WD, Pittelkow MR, Winkelmann RK. Clinical and pathologic correlations in 96 patients with panniculitis, including 15 patients with deficient levels of α 1-antitrypsin. Journal of the American Academy of Dermatology. 1989 Dec 31;21(6):1192-6. PubMed
- Valverde R, Rosales B, Ortiz-de Frutos FJ, Rodríguez-Peralto JL, Ortiz-Romero PL. Alpha-1-antitrypsin deficiency panniculitis. Dermatologic Clinics. 2008 Oct 31;26(4):447-51 PubMed
- Hendrick SJ, Silverman AK, Solomon AR, Headington JT. α 1-Antitrypsin deficiency associated with panniculitis. Journal of the American Academy of Dermatology. 1988 Apr 30;18(4):684-92. PubMed
- Geller JD, Su WD. A subtle clue to the histopathologic diagnosis of early α 1-antitrypsin deficiency panniculitis. Journal of the American Academy of Dermatology. 1994 Aug 31;31(2):241-5. PubMed
- Smith KC, Pittelkow MR, Su WD. Panniculitis Associated With Severe a1-Antitrypsin Deficiency: Treatment and Review of the Literature. Archives of Dermatology. 1987 Dec 1;123(12):1655-61. PubMed
- Pinto AR, Maciel LS, Carneiro F, Resende C, Chaves FC, Freitas AF. Systemic nodular panniculitis in a patient with alpha‐1 antitrypsin deficiency (PiSS phenotype). Clinical and Experimental Dermatology. 1993 Mar 1;18(2):154-5. PubMed
- Linares-Barrios M, Conejo-Mir IS, Artola Igarza JL, Navarrete M. Panniculitis due to α1‐antitrypsin deficiency induced by cryosurgery. British Journal of Dermatology. 1998 Mar 1;138(3):552-3. PubMed
- Pittelkow MR, Smith KC, Daniel SW. Alpha-1-antitrypsin deficiency and panniculitis: perspectives on disease relationship and replacement therapy. The American Journal of Medicine. 1988 Jun 24;84(6):80-6. PubMed
- Olson JM, Moore EC, Valasek MA, Williams LH, Vary JC. Panniculitis in alpha-1 antitrypsin deficiency treated with enzyme replacement. Journal of the American Academy of Dermatology. 2012 Apr 30;66(4):e139-41. PubMed
- Al-Niaimi F, Lyon C. Severe ulcerative panniculitis caused by alpha 1-antitrypsin deficiency: remission induced and maintained with intravenous alpha 1-antitrypsin. Journal of the American Academy of Dermatology. 2011 Jul 31;65(1):227-9. PubMed
- Chowdhury MM, Williams EJ, Morris JS, Ferguson BJ, McGregor AD, Hedges AR, Stamatakis JD, Pope FM. Severe panniculitis caused by homozygous ZZ α1‐antitrypsin deficiency treated successfully with human purified enzyme (Prolastin®). British Journal of Dermatology. 2002 Dec 1;147(6):1258-61. PubMed
On DermNet
Other websites
- Alpha-1 Foundation
- Learning About Alpha-1 Antitrypsin Deficiency (AATD)– National Human Genome Research Institute (USA)