
Pseudoxanthoma elasticum — extra information
Introduction Demographics Causes Clinical features Variation in skin types Complications Diagnosis Differential diagnoses Treatment Prevention Outcome
What is pseudoxanthoma elasticum?
Pseudoxanthoma elasticum (PXE) refers to a group of rare inherited disorders that affect the elastic connective tissue of the skin, eyes, and blood vessels.
PXE, alternatively called Gronblad-Strandberg syndrome, was first described in 1881 and occurs when abnormal calcification and mineralization of elastic fibres in affected tissues occur. It is characterised by yellow papules and plaques (which may mimic xanthomas), skin laxity, and ocular manifestations that may incur vision loss at later stages.
Who gets pseudoxanthoma elasticum?
PXE is an autosomal recessive disease. While the true epidemiology of PXE is not currently known, the following has been estimated:
- Prevalence ranges from 1 in 100,000 to 1 in 25,000 which translates to a heterozygous carrier frequency of 0.63–1.26%
- Women and Caucasians may have a higher prevalence
- Average age at diagnosis is 30–40 years; may vary depending on disease genotype.
What causes pseudoxanthoma elasticum?
PXE is caused by inactivating mutations in a gene called ABCC6. The disease is autosomal recessive, meaning both copies of the defective gene must be present for PXE to occur. Over 300 mutation variants have been identified. Two common variants account for half of all cases of PXE.
PXE involves progressive deposition of calcium and phosphorus on elastic fibres. Deposits accumulate in elastin-rich tissue such as the reticular dermis of the skin, Bruch’s membrane of the choroid, and tunica media/intima of arteries. Elastic fibres eventually become fragmented.
The pathophysiology of PXE is currently understood to be due to metabolic derangement. ABCC6 encodes an ATP-dependent efflux transporter that excretes inorganic phosphate (PPi), a strong inhibitor of mineralization. Research has shown that decreased levels of PPi in the blood due to loss-of-function mutations of ABCC6 likely drive abnormal calcification in PXE.
What are the clinical features of pseudoxanthoma elasticum?
Cutaneous features
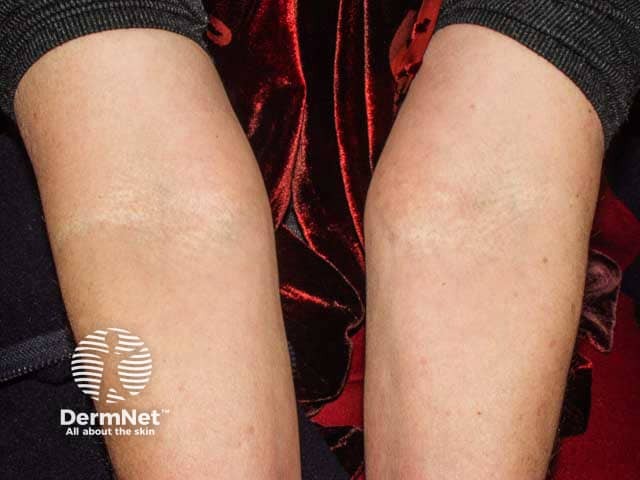
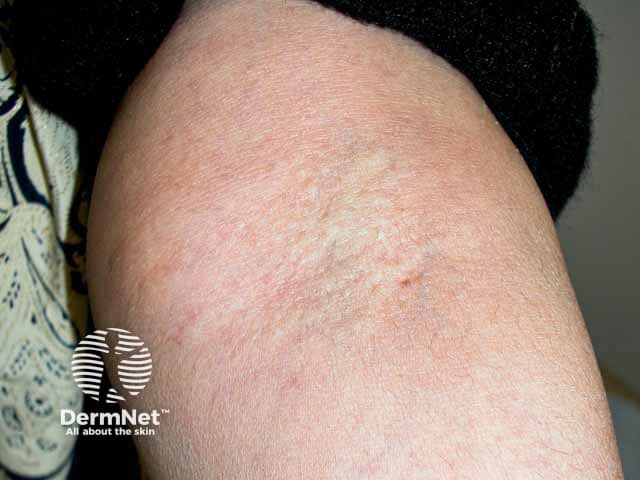
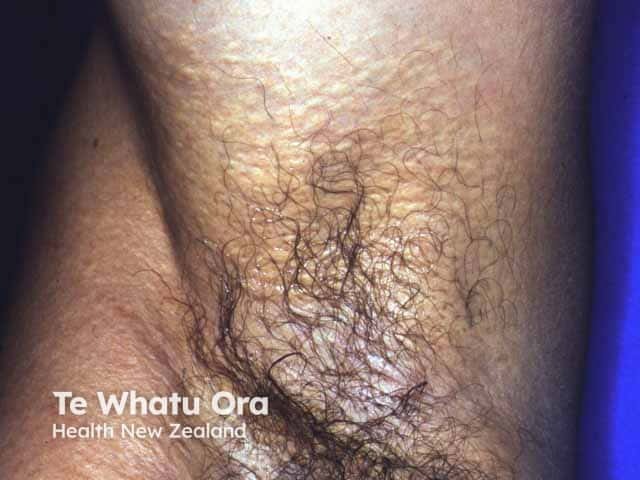
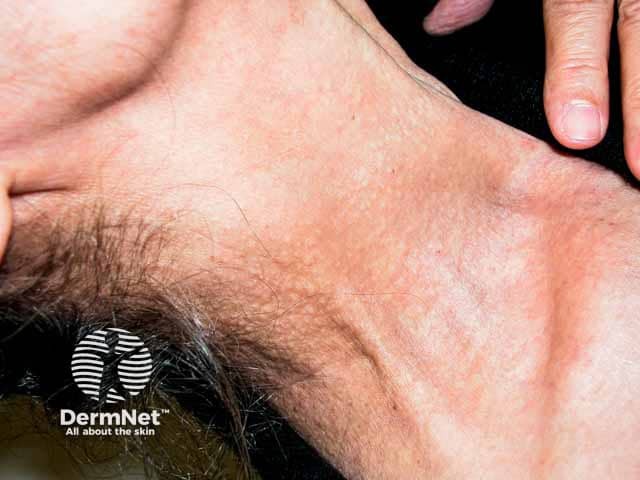
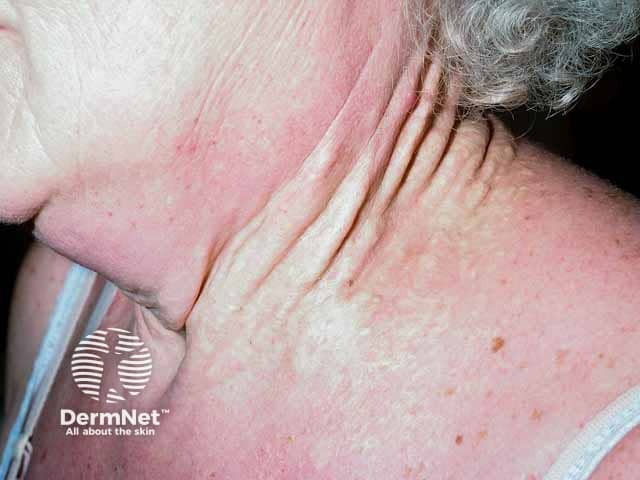
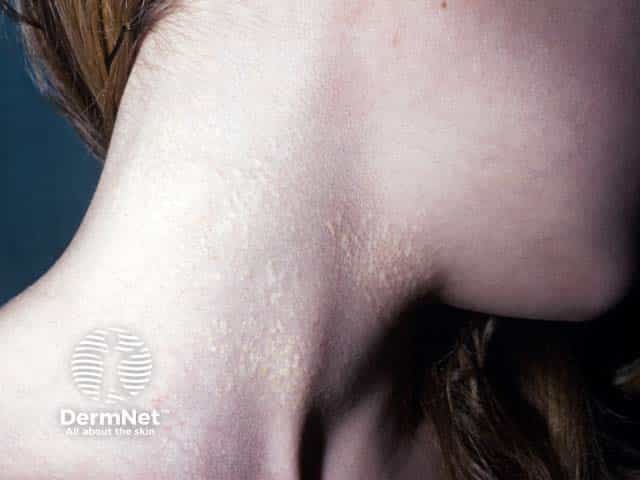
Skin is the first organ to be affected by PXE, beginning in childhood or adolescence. Findings include:
- 1–5 mm yellowish papules, distributed over the lateral neck and flexural areas
- Possible involvement of oral, rectal, or vaginal mucosa
- Reticular or linear arrangement of lesions
- Lesions progressively coalesce into larger plaques with a “cobblestone” appearance
- Affected skin becomes soft, lax, and slightly wrinkled over time
- Prominent mental (chin) fold
- Lesions are asymptomatic.
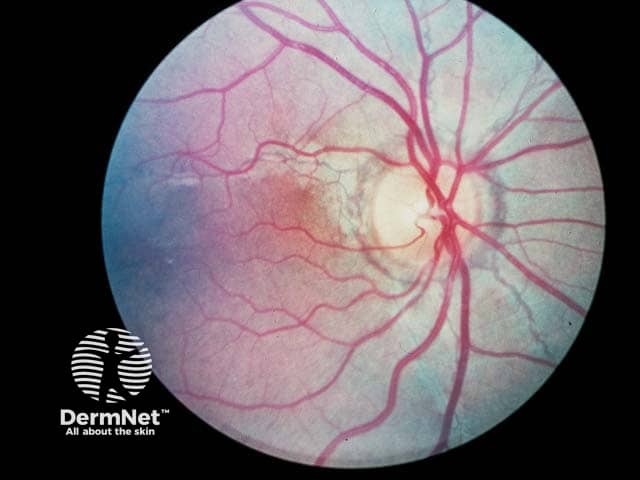
Ocular features
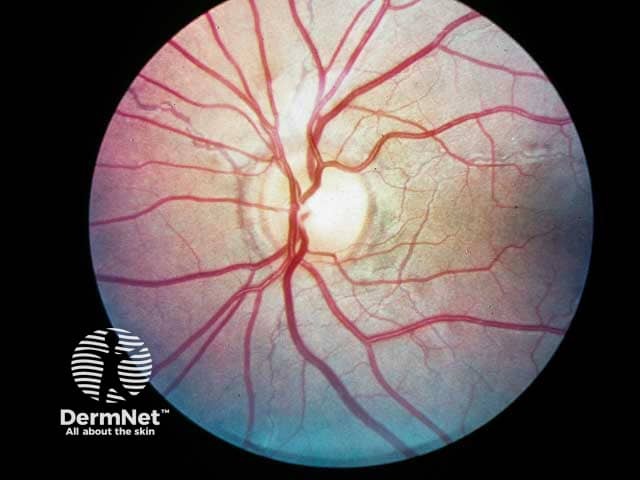
Bilateral ocular findings develop in all patients with PXE, beginning in childhood or early adolescence. Initial lesions are asymptomatic but may progress to vision loss with age.
- Peau d’orange (“skin of an orange”): mottled appearance of the temporal retina.
- Angioid streaks: narrow, irregular streaks that resemble blood vessels.
- “Comet tails'': areas of chorioretinal atrophy in the peripheral retina. These are a pathognomonic feature of PXE.
- Choroidal neovascularization (CNV): subretinal vessels growing through cracks in Bruch’s membrane. This is a major complication at later stages of disease that causes vision loss.
Ocular features of pseudoxanthoma elasticum
Cardiovascular features
- Peripheral artery disease
- Aneurysms
- Stroke (7–15%) and transient ischemic attacks
- Carotid stenosis
- Coronary artery disease at later stages, which may cause myocardial infarction and angina
Other systemic features
Other systemic features occasionally observed in PXE include:
- Gastrointestinal or urinary tract bleeding (15%)
- Benign calcification of breasts, pancreas, testicles, spleen, and liver
- Symptomatic calcification of renal arteries
- Reduced pulmonary function.
How do clinical features vary in differing types of skin?
Not much is known about the clinical features of PXE across different skin types. Certain mutation variants of ABCC6 have racial/ethnic associations and may cause varying disease phenotypes.
What are the complications of pseudoxanthoma elasticum?
- Vision loss
- Ischemic or haemorrhagic stroke
- Heart attack
- Severe limb claudication
- Acute GI bleeding
- Miscarriage is more common in early pregnancy
- Striae (stretch marks) are almost universal in pregnancy.
PXE is one of several conditions that can cause the skin condition elastosis perforans serpiginosa.
How is pseudoxanthoma elasticum diagnosed?
PXE may be diagnosed from its characteristic skin findings; histological confirmation is often useful. Ophthalmologists may perform a fundoscopy to diagnose pathognomonic ocular features.
Histopathology may be utilised in the diagnosis of PXE. Different staining techniques are used to reveal calcium deposits, shortened and fragmented elastic fibres, and deformation of collagen fibres.
Genetic testing for ABCC6 mutation homozygosity or compound heterozygosity is the gold standard test for PXE. Over 90% of patients have detectable mutations. Genetic testing can also rule out PXE-like conditions.
What is the differential diagnosis for pseudoxanthoma elasticum?
- Solar elastosis
- Acquired PXE (e.g. perforating calcific elastosis, chronic D-penicillamine use)
- Linear focal elastosis
- Mid-dermal elastolysis
- Cutis laxa
- Lesions resembling PXE can be seen in β-thalassemia and sickle cell disease.
What is the treatment for pseudoxanthoma elasticum?
The most important aspect of treatment is to ensure that complications from blood vessel involvement are prevented or dealt with speedily by the appropriate specialist.
General measures
- Reduction of cardiovascular risk factors (quitting smoking, exercise, weight loss, etc).
- Regular follow-up with cardiologists or vascular specialists as recommended.
- Regular eye exams.
Specific measures
There is no current treatment targeting the underlying disease process of PXE. However, treatments exist for various disease manifestations:
- Cosmetic surgery for skin laxity and lesions
- Intraocular vascular endothelial growth factor (VEGF) inhibitors, such as bevacizumab for CNV
- Vascular surgery for peripheral artery disease.
How do you prevent pseudoxanthoma elasticum?
PXE is not currently preventable but is a potential candidate for gene therapy. Genetic counselling may be helpful.
What is the outcome for pseudoxanthoma elasticum?
Individuals with PXE typically have a normal life expectancy. The extent of ocular or cardiovascular complications determines the morbidity and mortality of disease.
Bibliography
- Germain DP. Pseudoxanthoma elasticum. Orphanet J Rare Dis. 2017;12(1):85. doi:10.1186/s13023-017-0639-8. Journal
- Jansen RS, Küçükosmanoglu A, de Haas M, et al. ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc Natl Acad Sci U S A. 2013;110(50):20206–11. doi:10.1073/pnas.1319582110. Journal
- Legrand A, Cornez L, Samkari W, et al. Mutation spectrum in the ABCC6 gene and genotype-phenotype correlations in a French cohort with pseudoxanthoma elasticum. Genet Med. 2017;19(8):909–17. doi:10.1038/gim.2016.213. Journal
- Uitto J, Jiang Q, Váradi A, Bercovitch LG, Terry SF. Pseudoxanthoma elasticum: diagnostic features, classification and treatment options. Expert Opinion on Orphan Drugs. 2014;2(6):567–77. doi:10.1517/21678707.2014.908702. PubMed Central
On DermNet
- Pseudoxanthoma elasticum pathology
- Pseudoxanthoma elasticum images
- Calcinosis cutis
- Elastosis
- Skin signs of gastrointestinal disease
Other websites
- PXE International
- The PXE Support Group — PIXiE (UK)
- National Assn for Pseudoxanthoma Elasticum Inc. (NAPE)
- Connective Tissue — geneSkin